For an introduction to some of our interests, check out the following publications:
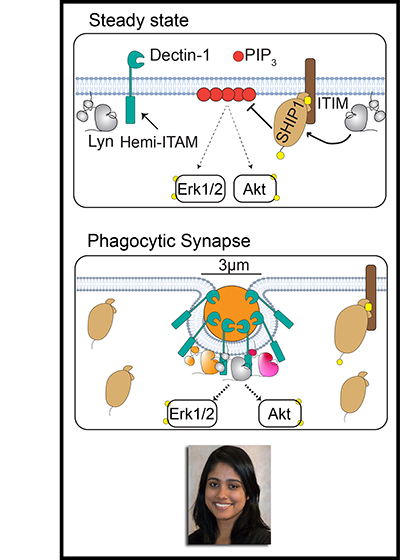
 Steady-state phosphorylation of SHIP1 by Lyn restricts macrophage activation in the absence of a phagocytic synapse
Steady-state phosphorylation of SHIP1 by Lyn restricts macrophage activation in the absence of a phagocytic synapse
Senevirathne, Kanagy, Cattley, Swanson, Toledo Ramos, Hawse, Freedman
(2025) bioRxiv. https://www.biorxiv.org/content/10.1101/2025.04.16.648985v1. PMID:TBD
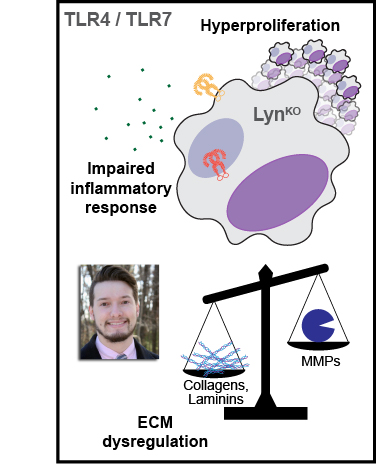
Lyn expression in macrophages promotes TLR activation and restricts proliferation in an isoform-independent manner
Lindstedt, Greene, Xu, Williams, Freedman
(2025) J Leukoc Biol. qiaf140. PMID:41054812
A dominant function of LynB kinase in preventing autoimmunity
Brian*, Sauer*, Greene* (co-first), Senevirathne, Lindstedt, Funk, Ruis, Ramirez, Auger, Swanson, Nunez, Moriarity, Lowell, Binstadt, Freedman
(2022) Sci Adv. 8(16):eabj5227. PMID:35452291
SH3-domain mutations selectively disrupt Csk homodimerization or PTPN22 binding
Brian, Sjaastad, Freedman (2022) Sci Rep. 12:5875. PMID:35393453
Regulation of myeloid-cell activation (review)
Greene, Brian, Senevirathne, Freedman (2021) Curr Opin Immunol. 73:34-42. PMID:34601225
The Src-family kinase Lyn in immunoreceptor signaling (review)
Brian, Freedman (2021) Endocrinology. 162(10):bqab152. PMID:34320188
Complete publication list:
Microscale engagement of the hemi-ITAM-containing receptor Dectin-1 by a fungus-derived particle initiates signaling through the Src-family kinases (SFKs) and Syk that leads to downstream Erk and Akt activation and the macrophage antimicrobial response. To minimize local tissue damage in the absence of a true pathogenic threat, macrophages must remain unresponsive to low-valency receptor ligation by soluble ligands from food or remote infection sites. To investigate how SFKs regulate macrophage sensitivity, we compared signaling in murine bone-marrow-derived macrophages (BMDMs) exposed to depleted zymosan, a particulate and highly multivalent Dectin-1 ligand, with signaling after pharmacologically induced SFK activation without receptor engagement or formation of a phagocytic synapse. We show that high-valency Dectin-1 engagement and formation of a phagocytic synapse restrict phosphorylation of the ITIM-associated phosphatase SHIP1 and promote Erk and Akt signaling. In contrast, SFK activation in the absence of a phagocytic synapse induces phosphorylation of SHIP1 and leads to dampened activation of Erk and Akt. Whereas multiple SFKs are capable of phosphorylating SHIP1 in principle, the SFK Lyn functions uniquely in maintaining steady-state phosphorylation of SHIP1 and setting tonic and induced levels of Erk and Akt phosphorylation. Consequently, Lyn has a special role in suppressing the Akt pathway and maintaining Erk/Akt pathway balance. Interestingly, formation of a phagocytic synapse circumvents this requirement for Lyn to maintain Erk/Akt balance. These findings highlight the unique function of Lyn in maintaining macrophage steady-state signaling and limiting pro-inflammatory responses to disorganized pathway activation and soluble microbial components.
Toll-like receptor (TLR) signaling is vital for antimicrobial macrophage function, and its dysregulation is associated with diseases such as lupus, multiple sclerosis, pulmonary fibrosis, and cancer. The Src-family kinase Lyn may have net activating or inhibitory effects on TLR signaling, yet distinct functions of the Lyn splice variants LynA and LynB in TLR signaling have not been investigated. We used isoform-specific Lyn knockout mice (LynAKO and LynBKO) to interrogate the contribution of each isoform to TLR signaling in bone-marrow-derived macrophages. Bulk RNA sequencing and cytokine analyses revealed that complete Lyn deficiency (LynKO) dampened TLR4- and TLR7-induced inflammatory gene expression and production of tumor necrosis factor (TNF) but enhanced the expression of genes responsible for synthesizing the extracellular matrix and promoting proliferation. Despite reduced expression of total Lyn in single-isoform Lyn knockout BMDMs, expression of either LynA or LynB alone was sufficient to preserve a wild-type-like transcriptome at steady state and after treatment with the TLR7 agonist R848. However, LynAKO and LynBKO macrophages did have impaired TNF production in response to the TLR4 agonist lipopolysaccharide. Additionally, LynAKO and LynBKO macrophages were as hyperproliferative as LynKO cells. These data suggest that Lyn promotes macrophage activation in response to TLR signaling and restrains aberrant proliferation and matrix deposition in a dose-dependent rather than isoform-specific manner.
Maureen McGargill, Beiyun Liu, Michael Kuhns, Daniel Mucida, Isabella Rauch, Lauren Rodda, Meghan Koch, Hugo Gonzalez, Ken Cadwell,
Tanya Freedman, Tiffany Scharschmidt, Richard Sever, Jose Ordovas-Montanes, Andrew Oberst, Brooke Runnette, Matthew Krummel (2025)
bioRxiv . doi: 10.1101/2025.10.31.685758.
PMID:TBD
Peer review serves as the cornerstone of scientific quality control. Yet, the current journal-centric system is hindered by long timelines, high publication costs, inconsistent review quality, systemic biases, and editorial gatekeeping. Notably, the system is built around misaligned measures of impact that are tethered to journal branding and conflate scientific rigor (Quality) with perceived significance (Impact). Here, we report findings from the Discovery Stack Pilot Study, which tested a scientist-designed, journal-independent peer review model. The Discovery Stack model integrates in-line reviewer comments to promote constructive, improvement-focused feedback and generates separate, multimodal assessments of scientific Quality and Impact. To examine its feasibility and effectiveness, manuscripts enrolled in the pilot were reviewed in parallel with traditional journal review. A total of 162 reviews were completed, and survey data from 86 participants were analyzed to evaluate the experience of both authors and reviewers. The results showed that reviewers effectively evaluated Quality and Impact as separate dimensions, with Quality scores being more consistent across reviewers than Impact scores. Importantly, participants strongly supported the core elements of the Discovery Stack model and expressed enthusiasm for its broader adoption to enhance transparency, efficiency, and value in peer review. Future studies will explore integrating this model into a digital platform for reviewing and curating scientific discoveries to improve the production and dissemination of high-quality research.

Anders Matson, Rob Hullsiek, Kate Dixon, Sam Wang,
Anders Lindstedt, Ryan Friess, Shee Kwan Phung,
Tanya Freedman, Martin Felices, Emily Truckenbrod, Jianming Wu, Jeffrey Miller, Bruce Walcheck (2024)
J Immunother Cancer. 12(7):e008959.
PMID:39053944
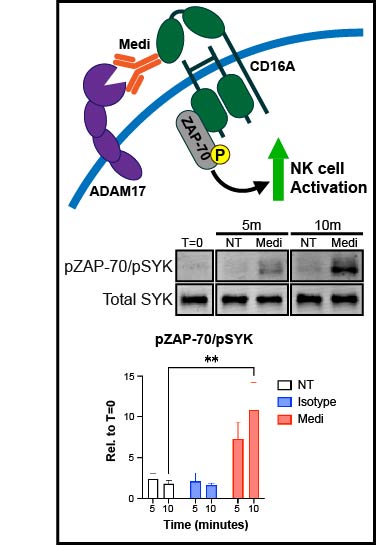
Background: Natural killer (NK) cells are being extensively studied as a cell therapy for cancer. These cells are activated by recognition of ligands and antigens on tumor cells. Cytokine therapies, such as IL-15, are also broadly used to stimulate endogenous and adoptively transferred NK cells in patients with cancer. These stimuli activate the membrane protease ADAM17, which cleaves various cell-surface receptors on NK cells as a negative feedback loop to limit their cytolytic function. ADAM17 inhibition can enhance IL-15-mediated NK cell proliferation in vitro and in vivo. In this study, we investigated the underlying mechanism of this process. Methods: Peripheral blood mononuclear cells (PBMCs) or enriched NK cells from human peripheral blood, either unlabeled or labeled with a cell proliferation dye, were cultured for up to 7 days in the presence of rhIL-15±an ADAM17 function-blocking antibody. Different fully human versions of the antibody were generated; Medi-1 (IgG1), Medi-4 (IgG4), Medi-PGLALA, Medi-F(ab')2, and TAB16 (anti-ADAM17 and anti-CD16 bispecific) to modulate CD16A binding. Flow cytometry was used to assess NK cell proliferation and phenotypic markers, immunoblotting to examine CD16A signaling, and IncuCyte-based live cell imaging to measure NK cell antitumor activity. Results: The ADAM17 function-blocking monoclonal antibody (mAb) Medi-1 markedly increased early NK cell activation by IL-15. By using different engineered versions of the antibody, we demonstrate involvement by CD16A, an activating Fcγ receptor and well-described ADAM17 substrate. Hence, Medi-1 when bound to ADAM17 on NK cells is engaged by CD16A and blocks its shedding, inducing and prolonging its signaling. This process did not promote evident NK cell fratricide or dysfunction. Synergistic signaling by Medi-1 and IL-15 enhanced the upregulation of CD137 on CD16A+ NK cells and augmented their proliferation in the presence of PBMC accessory cells or an anti-CD137 agonistic mAb.Conclusions: Our data reveal for the first time that CD16A and CD137 underpin Medi-1 enhancement of IL-15-driven NK cell activation and proliferation, respectively, with the latter requiring PBMC accessory cells. The use of Medi-1 represents a novel strategy to enhance IL-15-driven NK cell proliferation, and it may be of therapeutic importance by increasing the antitumor activity of NK cells in patients with cancer.
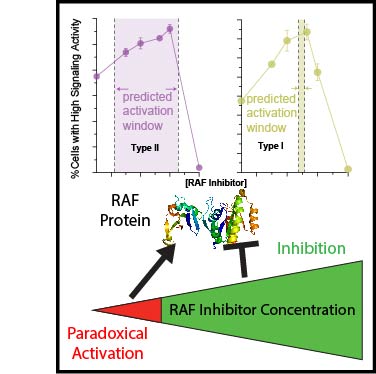
The type II class of RAF inhibitors currently in clinical trials paradoxically activate BRAF at subsaturating concentrations. Activation is mediated by induction of BRAF dimers, but why activation rather than inhibition occurs remains unclear. Using biophysical methods tracking BRAF dimerization and conformation we built an allosteric model of inhibitor-induced dimerization that resolves the allosteric contributions of inhibitor binding to the two active sites of the dimer, revealing key differences between type I and type II RAF inhibitors. For type II inhibitors the allosteric coupling between inhibitor binding and BRAF dimerization is distributed asymmetrically across the two dimer binding sites, with binding to the first site dominating the allostery. This asymmetry results in efficient and selective induction of dimers with one inhibited and one catalytically active subunit. Our allosteric models quantitatively account for paradoxical activation data measured for 11 RAF inhibitors. Unlike type II inhibitors, type I inhibitors lack allosteric asymmetry and do not activate BRAF homodimers. Finally, NMR data reveal that BRAF homodimers are dynamically asymmetric with only one of the subunits locked in the active αC-in state. This provides a structural mechanism for how binding of only a single αC-in inhibitor molecule can induce potent BRAF dimerization and activation.
Anna Jaeger, Chelsea Crooks, Andrea Weiler, Mason Bliss, Sierra Rybarczyk, Alex Richardson, Morgan Einwalter, Eric Peterson, Saverio Capuano, Alison Barkhymer, Jordan Becker,
J.T. Greene,
Tanya Freedman, Ryan Langlois, Thomas Friedrich, Matt Aliota (2023)
Sci Adv. 9(26):eadg3444.
PMID:37390207
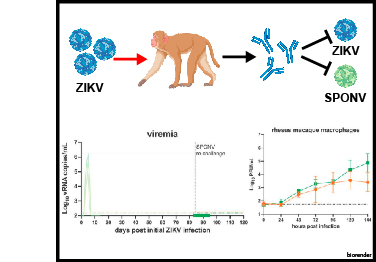
Spondweni virus (SPONV) is the closest known relative of Zika virus (ZIKV). SPONV pathogenesis resembles that of ZIKV in pregnant mice, and both viruses are transmitted by Aedes aegypti mosquitoes. We aimed to develop a translational model to further understand SPONV transmission and pathogenesis. We found that cynomolgus macaques (Macaca fascicularis) inoculated with ZIKV or SPONV were susceptible to ZIKV but resistant to SPONV infection. In contrast, rhesus macaques (Macaca mulatta) supported productive infection with both ZIKV and SPONV and developed robust neutralizing antibody responses. Crossover serial challenge in rhesus macaques revealed that SPONV immunity did not protect against ZIKV infection, whereas ZIKV immunity was fully protective against SPONV infection. These findings establish a viable model for future investigation into SPONV pathogenesis and suggest that the risk of SPONV emergence is low in areas with high ZIKV seroprevalence due to one-way cross-protection between ZIKV and SPONV.
Song Yi Bae, Hannah Bergom, Abderrahman Day,
J.T. Greene, Zoi Sychev, Gabrianne Larson, Eva Corey, Stephen Plymate,
Tanya Freedman, Justin Hwang, Justin Drake (2023)
Front Endocrinol. 14:1093332.
PMID:37065756
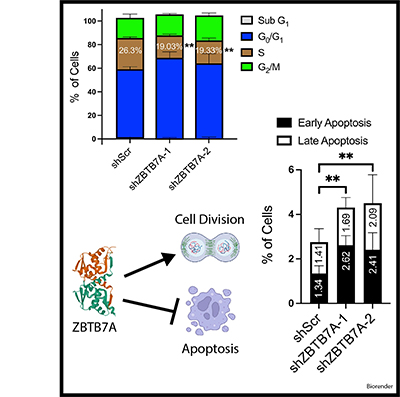
Neuroendocrine prostate cancer (NEPC) is a highly aggressive subtype of prostate cancer. NEPC is characterized by the loss of androgen receptor (AR) signaling and transdifferentiation toward small-cell neuroendocrine (SCN) phenotypes, which results in resistance to AR-targeted therapy. NEPC resembles other SCN carcinomas clinically, histologically and in gene expression. Here, we leveraged SCN phenotype scores of various cancer cell lines and gene depletion screens from the Cancer Dependency Map (DepMap) to identify vulnerabilities in NEPC. We discovered ZBTB7A, a transcription factor, as a candidate promoting the progression of NEPC. Cancer cells with high SCN phenotype scores showed a strong dependency on RET kinase activity with a high correlation between RET and ZBTB7A dependencies in these cells. Utilizing informatic modeling of whole transcriptome sequencing data from patient samples, we identified distinct gene networking patterns of ZBTB7A in NEPC versus prostate adenocarcinoma. Specifically, we observed a robust association of ZBTB7A with genes promoting cell cycle progression, including apoptosis regulating genes. Silencing ZBTB7A in a NEPC cell line confirmed the dependency on ZBTB7A for cell growth via suppression of the G1/S transition in the cell cycle and induction of apoptosis. Collectively, our results highlight the oncogenic function of ZBTB7A in NEPC and emphasize the value of ZBTB7A as a promising therapeutic strategy for targeting NEPC tumors.
Ben Brian*,
Monica Sauer*,
J.T. Greene* (co-first),
Erandika Senevirathne,
Anders Lindstedt,
Olivia Funk, Brian Ruis,
Luis Ramirez, Jennifer Auger,
Whitney Swanson,
Myra Nunez, Branden Moriarity, Clifford Lowell, Bryce Binstadt,
Tanya Freedman (2022)
Sci Adv. 8(16):eabj5227.
PMID:35452291
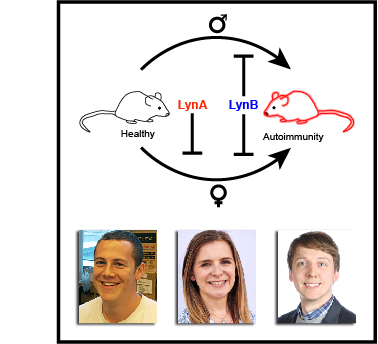
Here, we report that the LynB splice variant of the Src-family kinase Lyn exerts a dominant immunosuppressive function in vivo, whereas the LynA isoform is uniquely required to restrain autoimmunity in female mice. We used CRISPR-Cas9 gene editing to constrain lyn splicing and expression, generating single-isoform LynA knockout (LynAKO) or LynBKO mice. Autoimmune disease in total LynKO mice is characterized by production of antinuclear antibodies, glomerulonephritis, impaired B cell development, and overabundance of activated B cells and proinflammatory myeloid cells. Expression of LynA or LynB alone uncoupled the developmental phenotype from the autoimmune disease: B cell transitional populations were restored, but myeloid cells and differentiated B cells were dysregulated. These changes were isoform-specific, sexually dimorphic, and distinct from the complete LynKO. Despite the apparent differences in disease etiology and penetrance, loss of either LynA or LynB had the potential to induce severe autoimmune disease with parallels to human systemic lupus erythematosus (SLE).
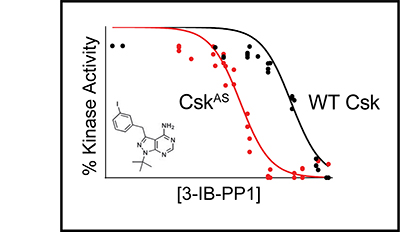
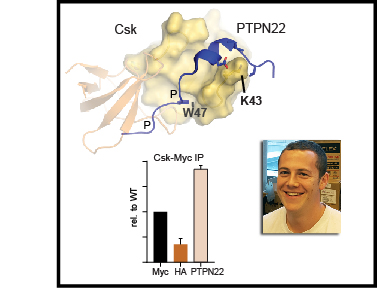
The kinase Csk is the primary negative regulator of the Src-family kinases (SFKs, e.g., Lck, Fyn, Lyn, Hck, Fgr, Blk, Yes), phosphorylating a tyrosine on the SFK C-terminal tail that mediates autoinhibition. Csk also binds phosphatases, including PTPN12 (PTP-PEST) and immune-cell PTPN22 (LYP/Pep), which dephosphorylate the SFK activation loop to promote autoinhibition. Csk-binding proteins (e.g., CBP/PAG1) oligomerize within membrane microdomains, and high local concentration promotes Csk function. Purifed Csk homodimerizes in solution through an interface that overlaps the phosphatase binding footprint. Here we demonstrate that Csk can homodimerize in Jurkat T cells, in competition with PTPN22 binding. We designed SH3-domain mutations in Csk that selectively impair homodimerization (H21I) or PTPN22 binding (K43D) and verifed their kinase activity in solution. Disruption of either interaction in cells, however, decreased the negative-regulatory function of Csk. Csk W47A, a substitution previously reported to block PTPN22 binding, had a secondary efect of impairing homodimerization. Csk H21I and K43D will be useful tools for dissecting the protein-specifc drivers of autoimmunity mediated by the human polymorphism PTPN22 R620W, which impairs interaction with Csk and with the E3 ubiquitin ligase TRAF3. Future investigations of Csk homodimer activity and phosphatase interactions may reveal new facets of SFK regulation in hematopoietic and non-hematopoietic cells.
Myeloid cells (macrophages, monocytes, dendritic cells, and granulocytes) survey the body for signs of infection and tissue damage and regulate tissue homeostasis, organogenesis, and immunity. They express receptors that initiate the inflammatory response, send signals that alter the vascular and cytokine milieu, and oversee the recruitment, differentiation, and activation of other myeloid and adaptive immune cells. Their activation must therefore be tightly regulated, optimized for maximal innate-immune protection with a minimum of collateral tissue damage or disorganization. In this review we discuss what it means for myeloid cells to become activated, with emphasis on the receptors and signaling molecules important for the recognition of pathogen- and damage-associated molecular patterns. We also outline how these signals are regulated by the steric properties of proteins, by adhesive and cytoskeletal interactions, and by negative feedback to keep inflammation in check and support healthy tissue development and homeostasis. Throughout the text we highlight recent publications and reviews that illustrate key elements of myeloid-cell regulation and direct readers therein for a comprehensive bibliography.
Effective regulation of immune-cell activation is critical for ensuring that the immune response, and inflammation generated for the purpose of pathogen elimination, are limited in space and time to minimize tissue damage. Autoimmune disease can occur when immunoreceptor signaling is dysregulated, leading to unrestrained inflammation and organ damage. Conversely, tumors can coopt the tissue healing and immunosuppressive functions of hematopoietic cells to promote metastasis and evade therapy. The Src-family kinase Lyn is an essential regulator of immunoreceptor signaling, initiating both proinflammatory and suppressive signaling pathways in myeloid immune cells (eg, neutrophils, dendritic cells, monocytes, macrophages) and in B lymphocytes. Defects in Lyn signaling are implicated in autoimmune disease, but mechanisms by which Lyn, expressed along with a battery of other Src-family kinases, may uniquely direct both positive and negative signaling remain incompletely defined. This review describes our current understanding of the activating and inhibitory contributions of Lyn to immunoreceptor signaling and how these processes contribute to myeloid and B-cell function. We also highlight recent work suggesting that the 2 proteins generated by alternative splicing of lyn, LynA and LynB, differentially regulate both immune and cancer-cell signaling. These principles may also extend to other Lyn-expressing cells, such as neuronal and endocrine cells. Unraveling the common and cell-specific aspects of Lyn function could lead to new approaches to therapeutically target dysregulated pathways in pathologies ranging from autoimmune and neurogenerative disease to cancer.
The response to the COVID-19 crisis across most research institutions mandated ceasing nonessential research activities in order to minimize the spread of the virus in our communities. With minimal notice, experiments were terminated, cell lines were frozen, mouse colonies were culled, and trainees were prevented from performing bench research. Still, despite the interruption of experimental productivity, the shutdown has proven for many PIs and trainees that doing and thinking science are not activities that are bound to the laboratory. Furthermore, the shutdowns have solidified important emerging trends and forced us to further innovate to get the most out of working remotely. We hope that some of these innovations, hard-gained in this difficult time, will persist and develop into new paradigms-- learnings that will improve our science and our relationship to climate and community beyond the current pandemic.
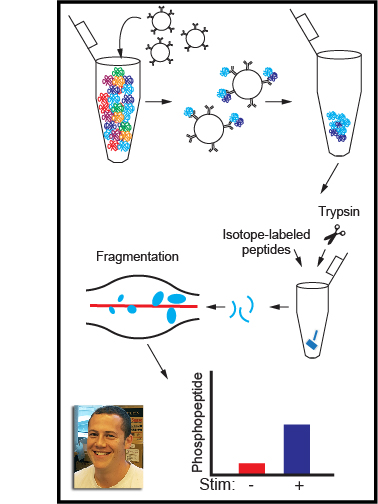
In this article we describe the use of pharmacological and genetic tools coupled with immunoblotting (Western blotting) and targeted mass spectrometry to quantify immune signaling and cell activation mediated by tyrosine kinases. Transfer of the ATP γ phosphate to a protein tyrosine residue activates signaling cascades regulating the differentiation, survival, and effector functions of all cells, with unique roles in immune antigen receptor, polarization, and other signaling pathways. Defining the substrates and scaffolding interactions of tyrosine kinases is critical for revealing and therapeutically manipulating mechanisms of immune regulation. Quantitative analysis of the amplitude and kinetics of these effects is becoming ever more accessible experimentally and increasingly important for predicting complex downstream effects of therapeutics and for building computational models. Secondarily, quantitative analysis is increasingly expected by reviewers and journal editors, and statistical analysis of biological replicates can bolster claims of experimental rigor and reproducibility. Here we outline methods for perturbing tyrosine kinase activity in cells and quantifying protein phosphorylation in lysates and immunoprecipitates. The immunoblotting techniques are a guide to probing the dynamics of protein abundance, protein–protein interactions, and changes in post-translational modification. Immunoprecipitated protein complexes can also be subjected to targeted mass spectrometry to probe novel sites of modification and multiply modified or understudied proteins that cannot be resolved by immunoblotting. Together, these protocols form a framework for identifying the unique contributions of tyrosine kinases to cell activation and elucidating the mechanisms governing immune cell regulation in health and disease.
Ben Brian,
Adrienne Jolicoeur, Candace Guerrero,
Myra Nunez, Zoi Sychev, Siv Hegre, Pål Sætrom, Nagy Habib, Justin Drake, Kathryn Schwertfeger,
Tanya Freedman (2019)
eLife. 8:e46043.
PMID:31282857
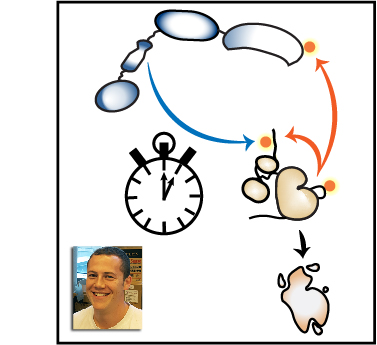
The activity of Src-family kinases (SFKs), which phosphorylate immunoreceptor tyrosine-based activation motifs (ITAMs), is a critical factor regulating myeloid-cell activation. We reported previously (Freedman et al., 2015) that the SFK LynA is uniquely susceptible to rapid ubiquitin-mediated degradation in macrophages, functioning as a rheostat regulating signaling. We now report the mechanism by which LynA is preferentially targeted for degradation and how cell specificity is built into the LynA rheostat. Using genetic, biochemical, and quantitative phosphopeptide analyses, we found that the E3 ubiquitin ligase c-Cbl preferentially targets LynA via a phosphorylated tyrosine (Y32) in its unique region. This distinct mode of c-Cbl recognition depresses steady-state expression of LynA in macrophages derived from mice. Mast cells, however, express little c-Cbl and have correspondingly high LynA. Upon activation, mast-cell LynA is not rapidly degraded, and SFK-mediated signaling is amplified relative to macrophages. Cell-specific c-Cbl expression thus builds cell specificity into the LynA checkpoint.
Clustering of receptors associated with immunoreceptor tyrosine-based activation motifs (ITAMs) initiates the macrophage antimicrobial response. ITAM receptors engage Src-family tyrosine kinases (SFKs) to initiate phagocytosis and macrophage activation. Macrophages also encounter nonpathogenic molecules that cluster receptors weakly, and must tune their sensitivity to avoid inappropriate responses. To investigate this response threshold, we compared signaling in the presence and absence of receptor clustering using a small-molecule inhibitor of Csk, which increased SFK activation and produced robust membrane-proximal signaling. Surprisingly, receptor-independent SFK activation led to a downstream signaling blockade associated with rapid degradation of the SFK LynA. Inflammatory priming of macrophages upregulated LynA and promoted receptor-independent signaling. In contrast, clustering the hemi-ITAM receptor Dectin-1 induced signaling that did not require LynA or inflammatory priming. Together, the basal-state signaling checkpoint regulated by LynA expression and degradation and the signaling reorganization initiated by receptor clustering allow cells to discriminate optimally between pathogens and non-pathogens.
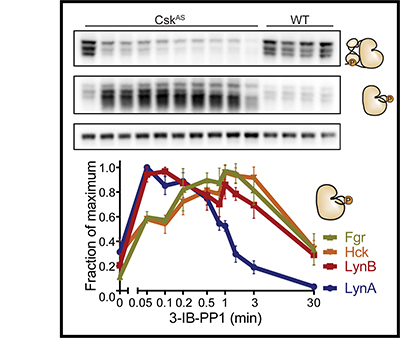
Signaling via the T cell antigen receptor (TCR) is initiated by Src-family kinases (SFKs). To understand how the kinase Csk, a negative regulator of SFKs, controls the basal state and the initiation of TCR signaling, we generated mice that express a Csk variant sensitive to an analog of the common kinase inhibitor PP1 (Csk(AS)). Inhibition of Csk(AS) in thymocytes, without engagement of the TCR, induced potent activation of SFKs and proximal TCR signaling up to phospholipase C-gamma1 (PLC-gamma1). Unexpectedly, increases in inositol phosphates, intracellular calcium and phosphorylation of the kinase Erk were impaired. Altering the actin cytoskeleton pharmacologically or providing costimulation via CD28 'rescued' those defects. Thus, Csk has a critical role in preventing TCR signaling. However, our studies also revealed a requirement for actin remodeling, initiated by costimulation, for full TCR signaling.
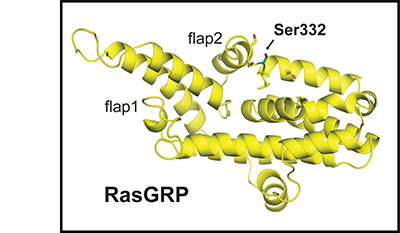
Clonal deletion of autoreactive B cells is crucial for the prevention of autoimmunity, but the signaling mechanisms that regulate this checkpoint remain undefined. Here we characterize a previously unrecognized Ca(2+)-driven pathway for activation of the kinase Erk, which was proapoptotic and biochemically distinct from Erk activation induced by diacylglycerol (DAG). This pathway required protein kinase C-δ (PKC-δ) and the guanine nucleotide-exchange factor RasGRP and depended on the concentration of the Ca(2+) sensor STIM1, which controls the magnitude of Ca(2+) entry. Developmental regulation of these proteins was associated with selective activation of the pathway in B cells prone to negative selection. This checkpoint was impaired in PKC-δ-deficient mice, which developed B cell autoimmunity. Conversely, overexpression of STIM1 conferred a competitive disadvantage to developing B cells. Our findings establish Ca(2+)-dependent Erk signaling as a critical proapoptotic pathway that mediates the negative selection of B cells.
ZAP-70 is a cytoplasmic protein tyrosine kinase that plays a critical role in the events involved in initiating T-cell responses by the antigen receptor. Here we review the structure of ZAP-70, its regulation, its role in development and in disease, and a model system in which ZAP-70 function can be interrupted by a chemical inhibitor. Our lab proposed a model by which the TCR initiates signaling via sequential interactions with cytoplasmic tyrosine kinases. When the TCR interacts with peptide antigen:MHC on antigen presenting cells, coreceptor-associated Lck is brought into proximity of the CD3 complex and phosphorylates ITAM tyrosines. When doubly phosphorylated, ITAMs recruit ZAP-70 via a high affinity interaction with the tandem SH2 domains of ZAP-70. This binding event leads to the release of ZAP-70 from its autoinhibited conformation, exposing the regulatory phosphorylation sites for Lck-mediated phosphorylation. Tyrosines in the activation loop of the ZAP-70 kinase domain are then phosphorylated by Lck or by ZAP-70 in trans to further promote its catalytic activity. A number of signaling proteins, including LAT and SLP-76, are subsequently phosphorylated by active ZAP-70. Phosphorylated LAT and SLP-76 function as scaffolds to recruit many other signaling molecules. These early signaling events eventually lead to T-cell activation, proliferation, and differentiation.
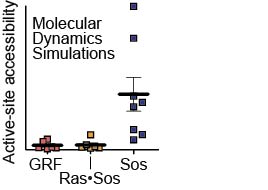
The Ras-specific nucleotide exchange factor Son of sevenless (Sos) is inactive without Ras bound to a distal allosteric site. In contrast, the catalytic domain of Ras guanine nucleotide releasing factor 1 (RasGRF1) is active intrinsically. By substituting residues from RasGRF1 into Sos, we have generated mutants of Sos with basal activity, partially relieved of their dependence on allosteric activation. We have performed molecular dynamics simulations showing how Ras binding to the allosteric site leads to a bias toward the active conformation of Sos. The trajectories show that Sos fluctuates between active and inactive conformations in the absence of Ras and that the activating mutations favor conformations of Sos that are more permissive to Ras binding at the catalytic site. In contrast, unliganded RasGRF1 fluctuates primarily among active conformations. Our results support the premise that the catalytic domain of Sos has evolved an allosteric activation mechanism that extends beyond the simple process of membrane recruitment.
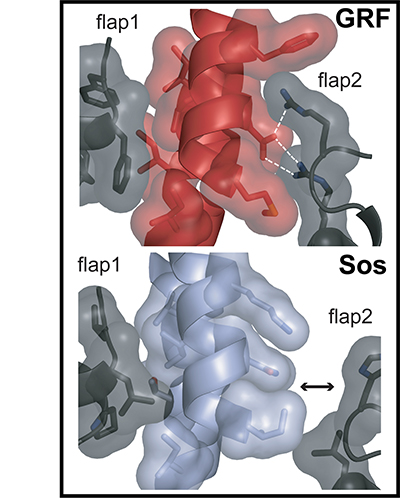
The Ras-specific guanine nucleotide-exchange factors Son of sevenless (Sos) and Ras guanine nucleotide-releasing factor 1 (RasGRF1) transduce extracellular stimuli into Ras activation by catalyzing the exchange of Ras-bound GDP for GTP. A truncated form of RasGRF1 containing only the core catalytic Cdc25 domain is sufficient for stimulating Ras nucleotide exchange, whereas the isolated Cdc25 domain of Sos is inactive. At a site distal to the catalytic site, nucleotide-bound Ras binds to Sos, making contacts with the Cdc25 domain and with a Ras exchanger motif (Rem) domain. This allosteric Ras binding stimulates nucleotide exchange by Sos, but the mechanism by which this stimulation occurs has not been defined. We present a crystal structure of the Rem and Cdc25 domains of Sos determined at 2.0-A resolution in the absence of Ras. Differences between this structure and that of Sos bound to two Ras molecules show that allosteric activation of Sos by Ras occurs through a rotation of the Rem domain that is coupled to a rotation of a helical hairpin at the Sos catalytic site. This motion relieves steric occlusion of the catalytic site, allowing substrate Ras binding and nucleotide exchange. A structure of the isolated RasGRF1 Cdc25 domain determined at 2.2-A resolution, combined with computational analyses, suggests that the Cdc25 domain of RasGRF1 is able to maintain an active conformation in isolation because the helical hairpin has strengthened interactions with the Cdc25 domain core. These results indicate that RasGRF1 lacks the allosteric activation switch that is crucial for Sos activity.
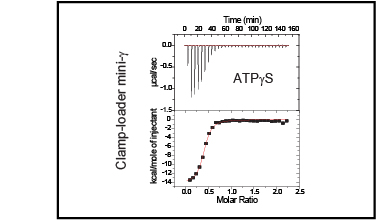
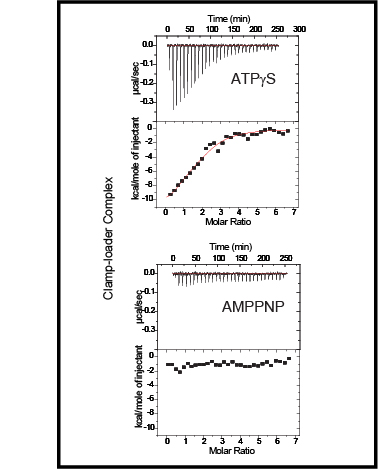
Clamp-loader complexes are heteropentameric AAA+ ATPases that load sliding clamps onto DNA. The structure of the nucleotide-free Escherichia coli clamp loader had been determined previously and led to the proposal that the clamp-loader cycles between an inactive state, in which the ATPase domains form a closed ring, and an active state that opens up to form a "C" shape. The crystal structure was interpreted as being closer to the active state than the inactive state. The crystal structure of a nucleotide-bound eukaryotic clamp loader [replication factor C (RFC)] revealed a different and more tightly packed spiral organization of the ATPase domains, raising questions about the significance of the conformation seen earlier for the bacterial clamp loader. We describe crystal structures of the E. coli clamp-loader complex bound to the ATP analog ATPgammaS (at a resolution of 3.5 A) and ADP (at a resolution of 4.1 A). These structures are similar to that of the nucleotide-free clamp-loader complex. Only two of the three functional ATP-binding sites are occupied by ATPgammaS or ADP in these structures, and the bound nucleotides make no interfacial contacts in the complex. These results, along with data from isothermal titration calorimetry, molecular dynamics simulations, and comparison with the RFC structure, suggest that the more open form of the E. coli clamp loader described earlier and in the present work corresponds to a stable inactive state of the clamp loader in which the ATPase domains are prevented from engaging the clamp in the highly cooperative manner seen in the fully ATP-loaded RFC-clamp structure.
Sliding clamps are loaded onto DNA by ATP-driven clamp loader complexes. The structure of the E. coli clamp loader in a nucleotide-free state has been determined previously. We now report crystal structures of a truncated form of the isolated gamma-ATPase subunit, gamma(1-243), of the E. coli clamp loader, in nucleotide-free and bound forms. The gamma subunit adopts a defined conformation when empty, in which the nucleotide binding site is blocked. The binding of either ATPgammaS or ADP, which are shown to bind with equal affinity to gamma(1-243), induces a change in the relative orientation of the two domains such that nucleotides can be accommodated. This change would break one of the gamma:gamma interfaces seen in the empty clamp loader complex, and may represent one step in the activation process.