
Our Research (For Nonscientists)
Welcome
Follow us on twitter @tfreedmanlab. Contact us at tfreedma@umn.edu. ![]() .
.
Thanks for stopping by! As scientists, we are driven by the thrill of discovery and a desire to contribute to society. We get front row seats to appreciate the stunning beauty of our universe and the exquisitely controlled dances of the molecules and cells within our bodies. We are excited to discover how changes in the smallest proteins can affect how cells communicate with each other or transition from healthy to disease-enabling. Millions of years of evolution have left us with carefully balanced systems, cells, and bodies. Quirks or imperfections that alter these balances are key drivers of human disease.
Our laboratory studies the immune system, specifically how immune cells can transmit information received from the environment (outside the cell) to meaningful instructions inside the cell and, further, how they can then transmit this information to neighboring cells or organs. As part of this process, we are interested in how proteins change shape and communicate with each other to translate these received signals into a cellular action.
Of particular importance to ensuring an appropriate cellular action is the decision an immune cell must make when first encountering potentially triggering molecules in the environment. In response to a harmful pathogen or infection, an immune cell must turn on (activate) to ensure that the pathogen is destroyed in a timely manner.
However, if immune cells are activated too strongly or in response to something normal in the body, they can drive autoimmune disease. Conversely, cancer cells can change how immune cells receive signals, dampening an anti-tumor immune response. Immune cells in tumors may even help cancer cells proliferate and metastasize by supporting tumors, healing damage, and keeping anti-tumor immune cells away.
By defining the processes regulating immune-cell signaling and specific regulatory steps that go awry in cancer and autoimmune disease, we ultimately will be able to manipulate these signaling pathways with safer and more effective therapies. We also believe in the value of basic research. We scientists know that we can never predict the full value of our fundamental discoveries to scientists and society in the future.
Our work has been funded mostly by grants from the National Institutes of Health (NIH), including the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), the National Institute of Allergy and Infectious Diseases (NIAID), the National Institute of Allergy and Infectious Diseases (NIAID), and the Office of Autoimmune Disease Research within the Office of Research on Women's Health (OARD-ORWH). Thank you (the public) for supporting the NIH and for appreciating the value of knowledge, data, and research science!
While we rely on public funds from the NIH for our research efforts, we have also been grateful for support from foundations and university programs, including the Rheumatology Research Foundation, American Cancer Society, and the following entities at the University of Minnesota: the Department of Pharmacology, Center for Immunology, Masonic Cancer Center, Center for Autoimmune Diseases Research, and University of Minnesota Foundation.
Select Publications and Research Concepts
The following sections highlight specific topics in our research. If you scroll down, you can find general information about the immune cells and proteins we study, with links to actual scientific journal webpages and pdf files. Look near the top for our most recent work.
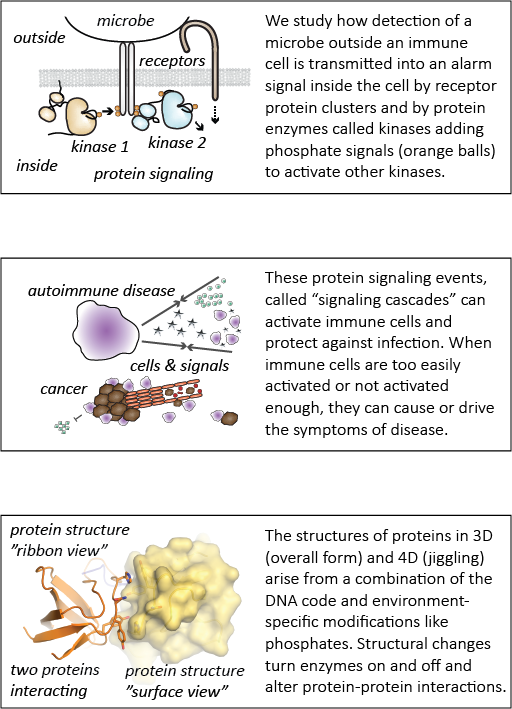
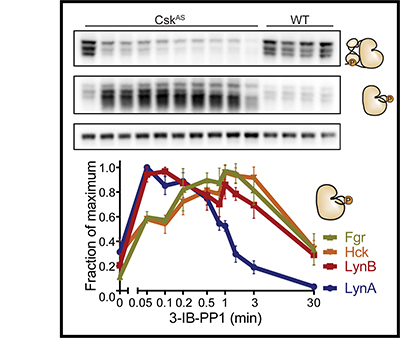
This paper describes the creation and description of two new mouse models for studying autoimmune disease. Previous research had shown that the Lyn acts as a brake on immune-cell activation and prevents mice from developing autoimmunity. However, previous research failed to account for the fact that that the gene that encodes Lyn actually makes two proteins – LynA and LynB. It wasn’t known whether LynA and LynB acted similarly or differently in preventing autoimmunity. Using a powerful gene-editing tool known as CRISPR-Cas9, we deleted LynA and LynB separately in mice to study the effects of losing either protein. We found that when LynB was deleted, both male and female mice developed autoimmunity. When LynA was deleted, only female mice got severe disease. Interestingly, the autoimmune disease that mice developed when either LynA or LynB was deleted looked differently than with both proteins were deleted together, suggesting that LynA and LynB could have non-overlapping roles in immune regulation. Further research using these mice will be valuable for studying why women are more likely to develop autoimmune disease than men. In the future, we hope to use this basic scientific knowledge to develop new approaches to treating these types of diseases.
This study was a tremendous group effort led by Ben Brian, Joseph (J.T.) Greene and Monica Sauer, with additional team efforts from others in the Freedman Lab (Erandika Senevirathne, Olivia Funk, Anders Lindstedt, Luis Ramirez, Whitney Swanson, and Myra Nunez). Our lab neighbors, Bryce Binstadt’s lab, provided critical help and the Mouse Genetics Laboratory and Genome Engineering Shared Resource at the University of Minnesota were instrumental in the creation of the new mouse models. Tanya (the principal investigator and senior author) secured funding and helped direct the project, analyze data, and write the paper.
(review article)
This review article was developed from a chapter of Ben Brian's PhD dissertation. Ben is now pursuing postdoctoral research in immunology at the University of California, Berkeley.
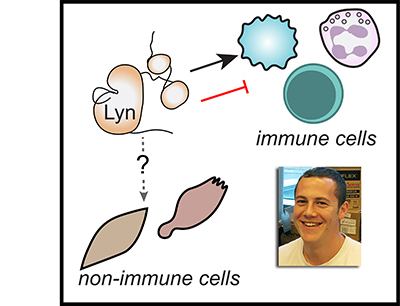
(viewpoint article)
This perspective piece was a true collaborative effort, incorporating the perspectives of all the authors above, faculty and trainees in Minnesota, San Francisco, and Seattle.

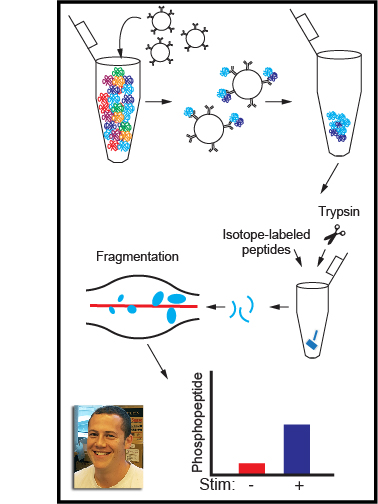
These step-by-step instructions describe methods for quantifying tyrosine kinase activation and tools to perturb tyrosine kinases in order to investigate how they regulate cellular activation. Tyrosine-kinase signaling is a critical component of immune-cell activation, and understanding the interactions of tyrosine kinases will hopefully lead to development of new medicines that alter immune cells to treat disease, including cancer and autoimmunity. These protocols can be divided into three basic sections. The first section describes our method for immunoblotting which is one of the workhorse methods in biochemistry for investigating the activation of proteins and protein phosphorylation. Section two describes a process for protein co-immunoprecipitation which is critical for understanding how proteins interact with each other. Finally, the third section describes a process for targeted mass spectrometry, a powerful technique allowing scientists to identify and characterize new phosphorylation sites.
There is still a lot we don’t really know about tyrosine-kinase activation and the proteins that interact with tyrosine kinases. We hope that by sharing these methods and discussing tools for manipulating kinase signaling we can provide a framework for other scientists studying kinase activation. The ultimate goal is to further our knowledge of tyrosine kinases to improve therapeutic targeting of immune cells in disease.
- Ben Brian (PhD student)
- Tanya Freedman, PhD (Principal Investigator)
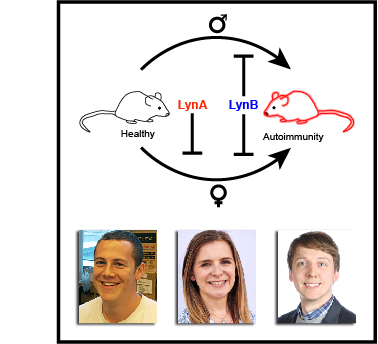
This paper describes how one of these germ sensors (a protein signaling pathway inside cells) is regulated in different cell types. Macrophages, which eat foreign cells like bacteria and fungi, express a protein called c-Cbl that helps prevent harmful inflammatory signaling. Mast cells, which help guard against parasitic infections, but are also implicated in allergic disease, do not express c-Cbl and thus can become activated very easily.
We have now discovered how the protein c-Cbl interacts with the LynA, a critical protein involved in inflammatory signaling, and the way this interaction controls macrophage-cell activation. A key modification of LynA, called phosphorylation, causes it to interact strongly with c-Cbl and is critical for controlling the strength of LynA signaling. This work shows a key difference between mast cells and macrophages. Mast cells make almost no c-Cbl protein, which allows them be activated rapidly in response to foreign invaders. This trigger-happy signaling can sometimes lead to problems like allergies and asthma. In contrast, macrophages make a lot of c-Cbl protein, and they are much more careful about when they become activated.
This study was mostly the work of Ben Brian, a PhD student. Adrienne and Myra in the Freedman lab did a few key experiments. Tanya (the principal investigator and senior author) secured funding and helped direct the project, analyze data, and write the paper. We had some important help from our friends in other labs at the University of Minnesota and from new technology given to us by collaborators in London and Norway.
- Ben Brian (PhD student)
- Tanya Freedman, PhD (Principal Investigator)
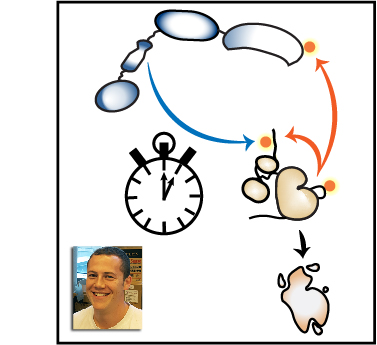
When the receptor proteins are activated by contact with a microbe, they interact with a family of proteins called SFKs, which in turn stimulate communication systems called signaling pathways inside the macrophages that activate their immune responses. However, these responses are only triggered if many receptors are activated and clustered together. Thus, macrophages are able to identify whether a cell is a normal part of the body or is a foreign invader by sensing the number of receptors that cluster together as they bind to the cell. However, it is not clear if it is the clustering itself that signals a genuine encounter with a microbe, or whether this information comes from the strength of the response by the SFK proteins.
We have developed a new experimental system that allows SFKs in macrophages to be directly activated without the need for receptor clustering. The experiments used macrophages obtained from genetically engineered mice and found that the direct activation of SFK proteins alone does not fully activate signaling pathways in the macrophages. A signaling blockade occurs due to the rapid destruction of an SFK protein called LynA. When the macrophages were exposed to molecules that are signals of inflammation in the body, they produced more LynA. This allowed these macrophages to be activated even without the formation of receptor clusters by interactions with pathogens.
Our findings reveal that LynA acts as a checkpoint, priming macrophages to respond more aggressively when the body is under attack. Future work will be aimed at understanding how LynA is destroyed and how the detection of real pathogens overcomes the checkpoint.
This study was conducted by Tanya Freedman when she was a postdoctoral researcher in the lab of Art Weiss at UCSF. She and Frankie Sjaastad (a researcher) finished the experiments and wrote the paper in her new lab at UMN. Ying, Kasia, and Boryana bred and handled the special mice that were essential for making this discovery. These animals were well cared for by special veterinarians. Clifford Lowell and Helen Goodridge were secondary mentors to Tanya and helped her learn to work with macrophage cells.
Review articles can be useful for scientists who are interested in learning a new field because they summarize the results of many scientific papers. Ideally, they also use this information to take a broad view of the field and contribute some new insight or understanding that could only be gained from such a bird's-eye view.
The authors of this article each contributed writing to different sections. Tanya Freedman was brought on as a contributor to the protein structure analyses in the review article and she worked with Byron Au-Yeung to make the figure. This figure was used as a cover image for the journal.
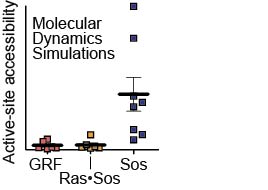
Tanya performed these computational and lab-bench experiments when she was a PhD student with John Kuriyan and Susan Marqusee at UC Berkeley. Holger, Olga, Tanja, and Greg also did some of the simulations and provided computational expertise. The movie to the right shows how the protein Sos jiggles over 5 microseconds in a computational experiment. The fixed rods show the active and inactive forms of the Sos protein, and the simulations show inactive Sos popping back and forth among the two structural states.
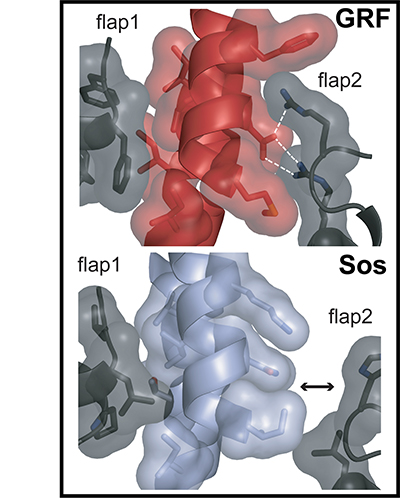
Ras exchange factors (including Sos and RasGRF1) are carefully regulated to prevent inappropriate activation of Ras. Although these proteins both bind to Ras and perform similar functions, they are regulated differently from each other. Sos is very hard to activate because the part of the protein that binds to Ras tends to fold up on itself and block interaction. When the active form of Sos is stabilized, though, Sos works very quickly to activate Ras. In contrast, RasGRF1 is a poor Ras activator but maintains an activated form without the need for this structural stabilization.
For this paper, we solved X-ray crystal structures (showing where the atoms are located and allowing us to build the cartoon models you can see in the illustration) and developed a scientific model explaining the functions of Sos and RasGRF1. The top panel of the illustration shows the structure of a Ras-binding part of RasGRF (red) with lots of contacts with flaps on either side of the same protein (black) holding it in an active position. The bottom panel shows the same part of inactive Sos (light blue), which is not held tightly by the flaps on either side and collapses down into an inactive position (tilting left instead of right, which blocks Ras binding).
Tanya and Holger obtained protein crystals of RasGRF1 and Sos, respectively, and solved the structures under the guidance of John Kuriyan and Susan Marqusee at UC Berkeley. Tanya also did some activity experiments. Tanja and Greg performed some computational experiments to help explain which parts of Sos and RasGRF1 contained important chemical differences.
Many thanks to Paulette Freedman for helping us get started in this outreach effort. Many of us are inexperienced at writing for a broad audience, and we appreciate the helping hand in setting the right tone and pitching our material at an appropriate level.